HTML
Short Communication - (2023) Volume 13, Issue 1
Technique to Execute Druggable Sites using Theoretical Proteins
Jessie Longaric**Correspondence: Jessie Longaric, Department of Analytical Chemistry, University of Yangon, Kamayut, Yangon, Myanmar, Email:
Received: 05-Dec-2022, Manuscript No. IJP-22-66730; Editor assigned: 07-Dec-2022, Pre QC No. IJP-22-66730(PQ); Reviewed: 26-Dec-2022, QC No. IJP-22-66730; Revised: 03-Jan-2023, Manuscript No. IJP-22-66730(R); Published: 07-Jan-2023, DOI: 10.37532/2249-1848.2023.13(1).30
Description
Protein structure is frequently investigated in order to better understand the elements that contribute to diseases, hereditary disorders, and nutritional makeup. In most research, Hypothetical Proteins (HPs) obtain recognition since they are frequently utilized in structural genomics with unclear activities due to a lack of evidence in vivo. If the sequence search does not match the protein that has been functionally described, the sequence is categorized as speculative. The hypothetical protein, according to Normi, is known as an orphan protein, yet it has been discovered that this HPs has a high potential for metal-to-protein trafficking and confers antibiotic resistance. Proteins are not self-contained; their functions are expressed when they interact with other molecules. Understanding the protein-ligand interaction is critical in functional annotation since it is key in drug discovery. The binding site prediction approach, which is separated into two methods: geometry and energy-based method, identifies the relationship of the protein-ligand based interaction. Binding site prediction software looks for a place that has the greatest chance of causing a binding contact with other molecules [1]. Data such as the binding pocket is provided by some predictors.
Antibiotic medication resistance is rising, necessitating the development of newer antimicrobial treatments. The identification of drug targets is the initial step in the drug discovery process. Traditional methods for identifying and characterizing microbial essential proteins require the use of compalex techniques such as high-throughput gene disruption systems, anti-sense RNA technology, allelic replacement mutagenesis, global transposon mutagenesis, systematic single-gene knockout experiments. Currently, functional genomics, comparative genomes, and bioinformatics give tools for better understanding pathogenic organisms and their host’s biological processes. These strategies could lead to the discovery of previously unknown cellular functions and pathways that could be used as therapeutic targets. For example, the Human Genome Project's completion has enabled a differential genomics strategy in which a subtractive dataset of a pathogen's genome and that of humans is created, with a listing of genes present in the pathogen but not in the human host.
The bioinformatics techniques also enable for the identification of a minimal set of genes required for a pathogen's survival. To automate the target identification procedure, several of these bioinformatics steps could be combined into a single in-silico tool [2,3]. The proteome subtraction approach, which is focused on identifying the intersection of a pathogen's proteome dataset with the host proteome to generate a listing of proteins that exist in pathogen but lack homologs in humans, can be used to identify in-silico therapeutic targets. Another way is to start with the pathogen's minimal essential protein dataset, which contains a list of proteins that could be expected to interfere with the pathogen's survival and so could be used as possible therapeutic targets. Sequence similarity-based technique and protein-protein interaction network topology-based approach are two frequently utilized in-silico approaches for predicting pathogen's minimal essential protein dataset.
Several in-silico techniques for identifying therapeutic targets in pathogenic organisms have been developed in the recent decade. The created a simple but effective computational technique to find pathogen proteins that are conserved among bacteria species but lack homologs in the yeast proteome. Based on in-silico differential genome analysis, created a method called 'Find Target.' Based on a comparison of bacterial proteomes, this tool assists users in searching for proteins that are present in one group of bacteria but not in another. The technique called 'T-iDT' for identifying therapeutic targets in bacteria; this programme uses a database of essential genes and a human proteome database to detect bacterial proteins that are non-homologs to human and are essential bacterial genes [4]. However, these methods lack the ability to identify and remove pathogen paralogous sequences, as well as the ability to remove very short pathogen sequences prior to homology mapping and the ability to update the host and essential protein databases.
Conclusion
In-silico subtractive genomics offers a rapid, powerful, and cost-effective technique to screen therapeutic and vaccine targets for any pathogen if both the pathogen and host genomes are available. However, experimental validation of the selected targets is required. This method necessitates several analyses at various phases. Similarly, an efficient integrated platform for doing the full analysis at the same time must be built. An approach independent of differentially expressed gene essential gene screening is also required. In such investigations, conserved essential genes based on pan genomics could be used as targets. To improve the efficacy of the original strategy, an in-silico mutagenesis approach and other computational validation approaches could be included in the analysis.
References
- Bencurova E, Gupta SK, Sarukhanyan E, et al. J Fungus. 2018;4(3):81.[CrossRef][Google Scholar][PubMed]
- Agamah FE, Mazandu GK, Hassan R, et al. Brief Bioinform.. 2020;21(5):1663-1675. [CrossRef][Google Scholar][PubMed]
- Chetouani F, Glaser P, Kunst F. Microbiology. 2001;147(10):2643-2649.[CrossRef][Google Scholar][PubMed]
- Singh NK, Selvam SM, Chakravarthy P. In-Silico Biol. 2006;6(6):485-493.[Google Scholar][PubMed]
Manuscript Submission
Submit your manuscript at Online Submission System
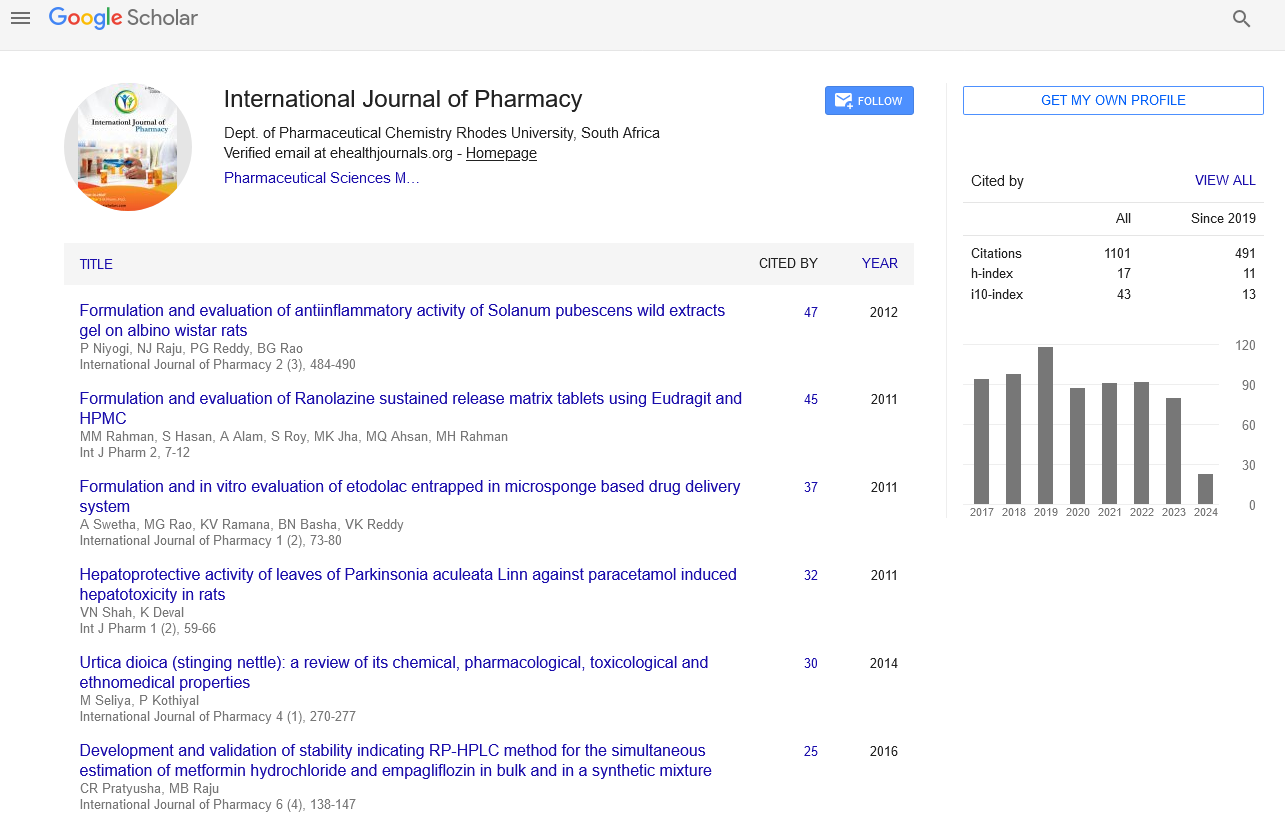
Google scholar citation report
Citations : 1101
International Journal of Pharmacy received 1101 citations as per google scholar report
International Journal of Pharmacy peer review process verified at publons
Indexed In
- CAS Source Index (CASSI)
- HINARI
- Index Copernicus
- Google Scholar
- The Global Impact Factor (GIF)
- Polish Scholarly Bibliography (PBN)
- Cosmos IF
- Open Academic Journals Index (OAJI)
- Directory of Research Journal Indexing (DRJI)
- EBSCO A-Z
- OCLC- WorldCat
- MIAR
- International committee of medical journals editors (ICMJE)
- Scientific Indexing Services (SIS)
- Scientific Journal Impact Factor (SJIF)
- Euro Pub
- Eurasian Scientific Journal Index
- Root indexing
- International Institute of Organized Research
- InfoBase Index
- International Innovative Journal Impact Factor
- J-Gate