HTML
Perspective Article - (2022) Volume 12, Issue 1
A Short Note on Pan-Genome Analyses
John Mark**Correspondence: John Mark, Department of Pharmacy, Central Philippine University, Iloilo, Philippines, Email:
Received: 07-Feb-2022, Manuscript No. IJP-22-154; Editor assigned: 11-Feb-2022, Pre QC No. IJP-22-154 (PQ); Reviewed: 21-Feb-2022, QC No. IJP-22-154; Revised: 28-Feb-2022, Manuscript No. IJP-22-154 (R); Published: 07-Mar-2022, DOI: 10.37532/2249-1848-22.12.05
Description
To account for intraspecific heterogeneity, prokaryotes proposed the notion of a species' pan-genome, which is the union of all 'core' conserved genes and all 'accessory' non-conserved genes across all strains of a species. Prokaryotes have been widely investigated in terms of species pan-genomes, but evidence of species pan-genomes has also been found in eukaryotes including plants and fungi. We analysed the pan-genomes of four model fungal species: Saccharomyces cerevisiae, Candida albicans, Cryptococcus neoformans var. grubii, and Aspergillus fumigatus, using a previously published methodology based on sequence homology and conserved microsynteny, as well as customized workflows.
In each of these species, between 80% and 90% of gene models are core genes that are highly conserved across all strains, many of which are involved in conserved survival mechanisms. The remaining 'accessory' gene models in several of these species are grouped inside subterminal areas and may have a role in disease and antibiotic resistance. The history of species core and accessory genomes shows that rather than wide-scale horizontal gene transfer, fungal pan-genomes evolve through strain-level innovations such gene duplication. The findings add to the growing body of evidence that eukaryote taxa have species pan-genomes.
Many branches of eukaryote functional and comparative genomics rely on curated reference genomes that are meant to be broadly representative of a species. Due to genetic and genomic heterogeneity across individuals within a species, reference genomes do not and cannot contain all genetic information for a species, regardless of their quality. To account for this variance, species with multiple genomes sequenced are frequently referred to as having a 'pan-genome,' which is defined as the union of all genes found across all isolates/strains of a species. After that, a species' pan-genome is usually separated into two halves.
(i) The 'core' genome, which contains genes that are conserved across all genomes of a species. These genes are usually, but not always, required for an individual organism's survival.
(ii) The 'accessory' or 'dispensable' genome, which contains genes unique to a species's isolate genomes or individual isolate genomes. Antibiotic-resistant and antibiotic-susceptible isolates of the same species may have diverse accessory genomes, indicating that these genes may impact phenotypic differences between isolates.
The pan-genome of a species can change as a result of its lifestyle: Sympatric species may have big pan-genomes (and consequently a lot of intraspecific variation), whereas ecologically isolated or highly specialised species have smaller pan-genomes.
The existence of a species pan-genome in prokaryotes was first demonstrated in 2005 across eight pathogenic strains of Streptococcus agalactiae, and was quickly confirmed by similar analyses of exemplar bacteria and archaea, such as Haemophilus influenzae, Escherichia coli and Sulfolobus islandicus. In recent years, a slew of tools for pangenome analysis have been released, utilising approaches including whole-genome alignment, read mapping, clustering algorithms, and de Bruijn graph creation.
Although the notion of the species pan-genome is well-established in comparative prokaryote genomics, it has only recently been applied to intraspecific comparative eukaryote genomics.
This is despite the fact that intraspecific genomic content variation has been observed in eukaryotes since the mid-2000s, when the first intraspecific comparative analysis of Saccharomyces cerevisiae genomes was performed. The relative difficulty of sequencing and analysing big eukaryotic genome datasets compared to prokaryotes is one reason for the lack of eukaryotic pan-genome analysis in the literature.
Furthermore, while Horizontal Gene Transfer (HGT) is assumed to be the primary force behind bacterial gene family and pan-genome evolution, HGT happens at much lower rates in eukaryotes and is much more difficult to detect. Despite these obstacles, a number of recent investigations of intraspecific variation in a variety of eukaryote taxa have found substantial support for the presence of a pan-genome in some form.
For example, a comparison of nine Brassica oleracea cultivars revealed that 19% of all genes were determined to be part of the B. oleracea accessory genome, with 2% of these being cultivarspecific. In the absence of rampant HGT, the techniques of pangenome evolution within eukaryotes appear to vary per species, and can include genome rearrangement events or more discrete adaptive evolution processes. Accessory genomes may emerge in plants as a result of ploidy, heterozygozity, and whole-genome duplication within species, as well as adaptive alterations and the evolution of phenotypic variations, as seen in B. oleracea. Adaptive evolution has also influenced the evolution of the Emiliania huxleyi pan-genome, with specialization resulting in strains with varied degrees of nutrient uptake and metabolism. Within the pan-genome of the Z. tritici species, there is a lot of functionally redundant accessory genome content, which is thought to come from the species' own genome defence mechanisms inducing polymorphisms rather than gene duplication events. A lot of accessory genes in Saccharomyces cerevisiae appear to have come from introgression from closely related Saccharomyces species, with a smaller number originating from HGT events with other yeasts.
Manuscript Submission
Submit your manuscript at Online Submission System
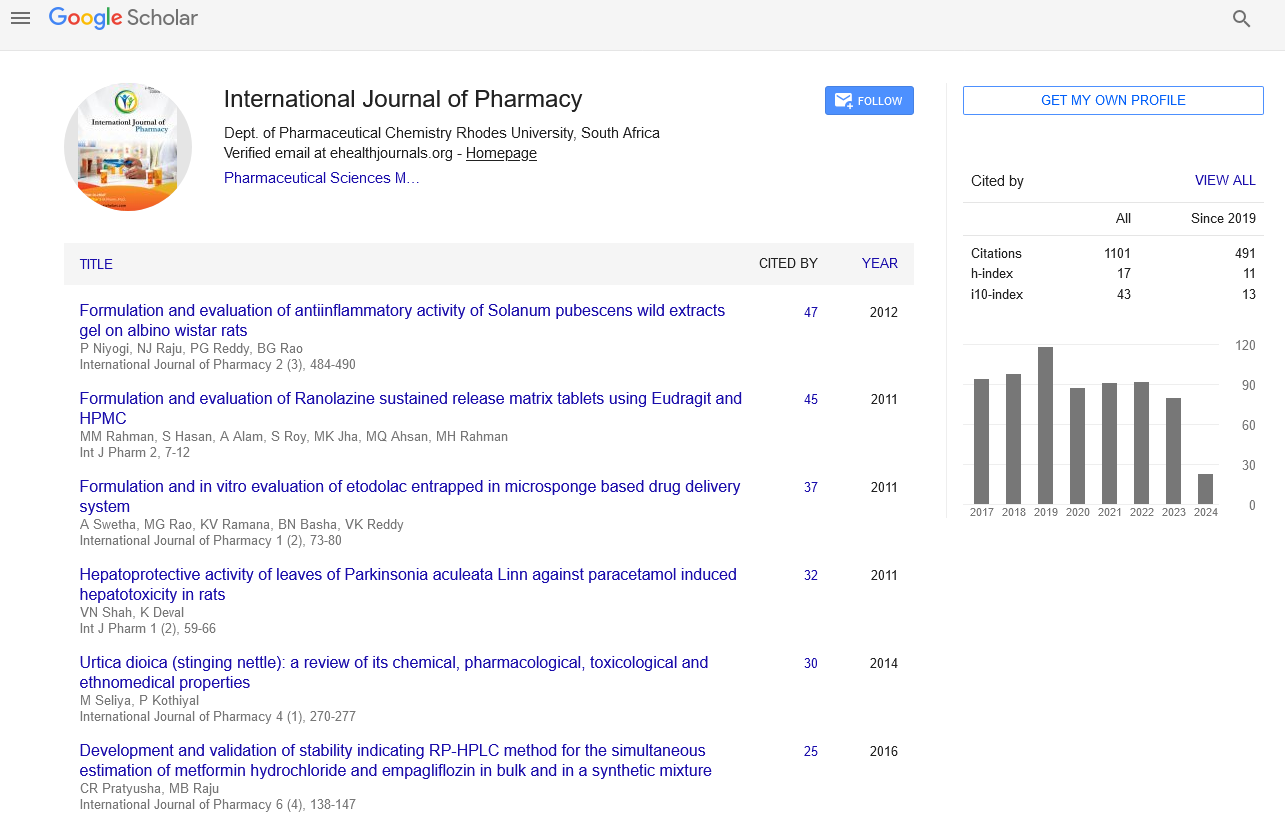
Google scholar citation report
Citations : 1101
International Journal of Pharmacy received 1101 citations as per google scholar report
International Journal of Pharmacy peer review process verified at publons
Indexed In
- CAS Source Index (CASSI)
- HINARI
- Index Copernicus
- Google Scholar
- The Global Impact Factor (GIF)
- Polish Scholarly Bibliography (PBN)
- Cosmos IF
- Open Academic Journals Index (OAJI)
- Directory of Research Journal Indexing (DRJI)
- EBSCO A-Z
- OCLC- WorldCat
- MIAR
- International committee of medical journals editors (ICMJE)
- Scientific Indexing Services (SIS)
- Scientific Journal Impact Factor (SJIF)
- Euro Pub
- Eurasian Scientific Journal Index
- Root indexing
- International Institute of Organized Research
- InfoBase Index
- International Innovative Journal Impact Factor
- J-Gate