Abstract
DEVELOPMENT AND VALIDATION OF RP-HPLC METHOD FOR THE ESTIMATION OF PROCESS RELATED IMPURITIES FROM NIMODIPINE BULK AND FORMULATION
Kasture V.S.*, Pawar S.S., Patil P.P., Musmade D.S., Ajage R. K. and Gehlot Ganjendra
The process related impurity of Nimodipine diethyl 1, 4-dihydro-2, 6-dimethyl pyridine 3, 5 dicarboxylate in bulk and formulations was synthesized, characterized and the RP-HPLC method was developed according to ICH Q2B guidelines for quantitation of impurity in bulk and formulations. The synthesis of intermediate was carried out by Hantzch process using m-nitrobenzaldehyde, ethylacetoacetate, in presence of ammonia and methanol as catalyst. The percentage yield was found to be 75%. The impurity was recrystallized and purified. The preliminary evaluation was done on lab scale viz. melting point, TLC and elemental analysis. The melting point of impurity was found to be 1560C. The TLC of impurity was carried by using Benzene and Methanol (6:1) and the Rf was found to be 0.80. The confirmation of structure of synthesized impurity was carried out by using sophisticated instrument viz, FT-IR, NMR, GC-MS etc. Finally, the RP-HPLC method was developed to identify and quantify the impurity in Nimodipine bulk and formulation as per ICH Q2B guidelines. The method was validated as per ICH guidelines. The method was found to be linear, precise, accurate, robust and rugged. Finally diethyl 1,4-dihydro-2,6-dimethyl pyridine 3,5 dicarboxylate impurity was quantified from bulk Nimodipine and its marketed tablet formulation. It was revealed that the amount of impurity present in tablet batch I and II was found to be 0.28% and 0.33% respectively and the bulk was found to be negligible. As per the ICH limit the amount of impurity more than 0.1% indicates that the impurity found in tablet formulations is potential impurity.
Manuscript Submission
Submit your manuscript at Online Submission System
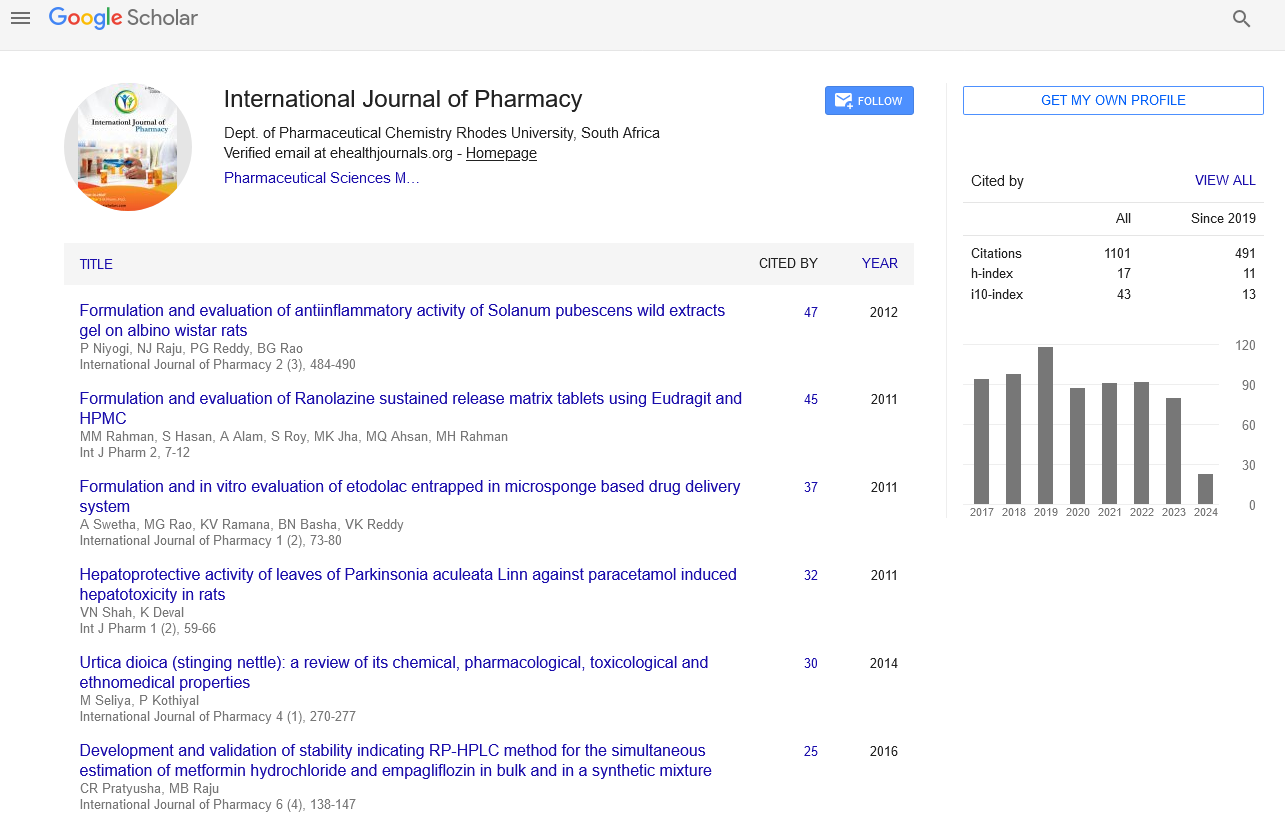
Google scholar citation report
Citations : 1101
International Journal of Pharmacy received 1101 citations as per google scholar report
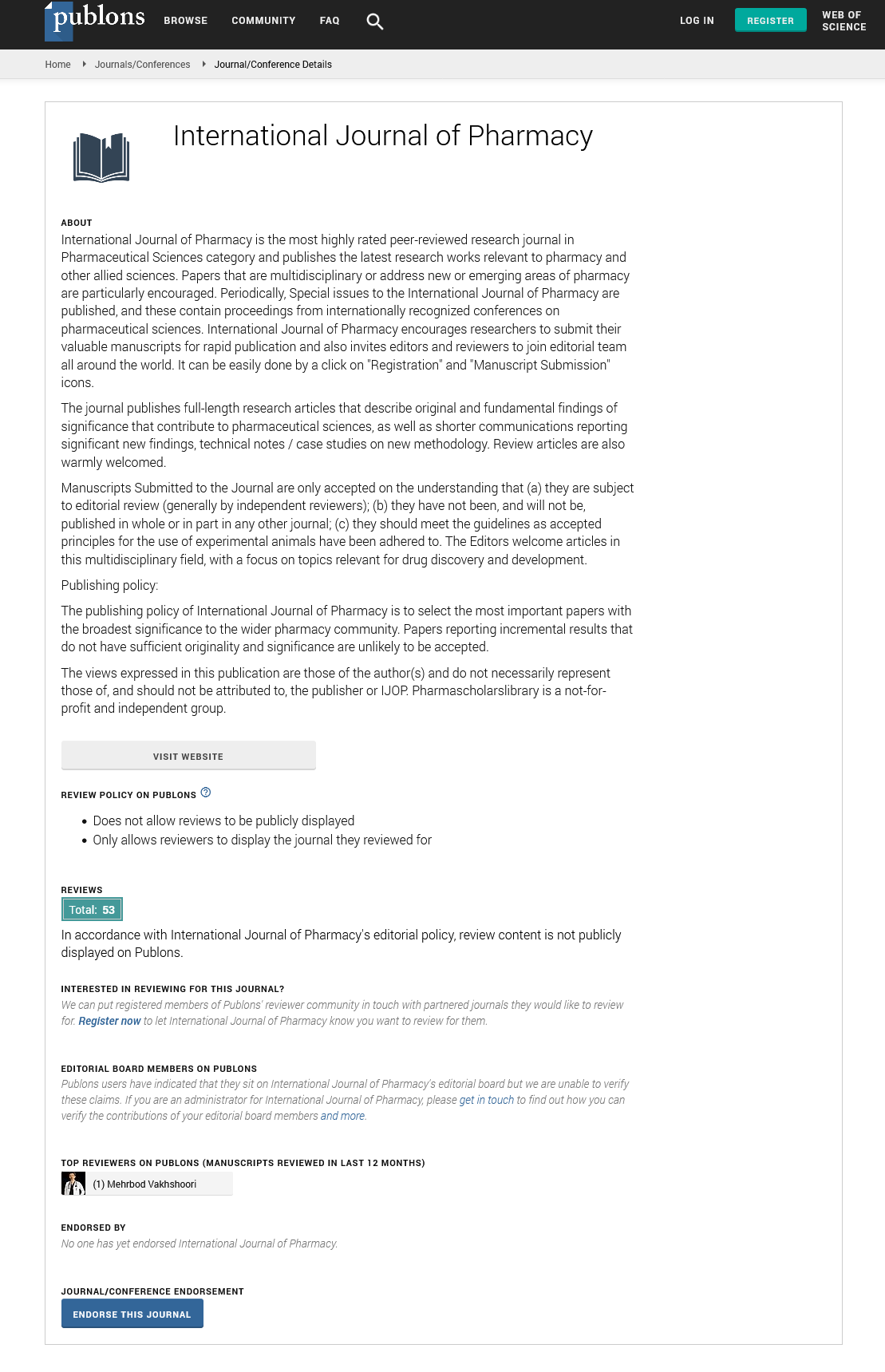
International Journal of Pharmacy peer review process verified at publons
Indexed In
- CAS Source Index (CASSI)
- HINARI
- Index Copernicus
- Google Scholar
- The Global Impact Factor (GIF)
- Polish Scholarly Bibliography (PBN)
- Cosmos IF
- Open Academic Journals Index (OAJI)
- Directory of Research Journal Indexing (DRJI)
- EBSCO A-Z
- OCLC- WorldCat
- MIAR
- International committee of medical journals editors (ICMJE)
- Scientific Indexing Services (SIS)
- Scientific Journal Impact Factor (SJIF)
- Euro Pub
- Eurasian Scientific Journal Index
- Root indexing
- International Institute of Organized Research
- InfoBase Index
- International Innovative Journal Impact Factor
- J-Gate